Interactomics
The interactome aims to study the main aspects of proteins interacting in a living system. The interactome is rather dynamic as the interactions and ultimately functions are manifested in a temporal and spatial manner. To understand the complex cellular mechanisms involved in a biological system, it is necessary to study the nature and specificity of these interactions and the dynamics involved in it at the molecular level, for which prediction of protein-protein interactions (PPIs) has played a significant role. My main research in interactomics focuses on machine learning algorithms for prediction and analysis of PPIs using low-throughput (mostly structural) and high-throughput data, with applications to calmodulin-binding proteins.
Prediction of Calmodulin-binding Proteins
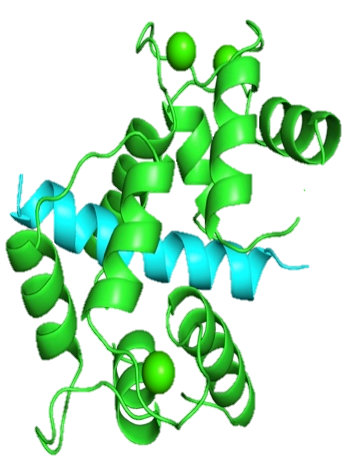 Calmodulin (CaM) is a calcium-binding protein that is a major transducer of calcium signaling and is a key signaling molecule for multicellular organisms. It has no enzymatic activity of its own but rather acts by binding to and altering the activity on a panel of cellular protein targets at a variety of motifs through binding mechanisms. Its targets are structurally and functionally diverse and participate in a wide range of physiological functions including immune response, muscle contraction and memory formation. Identifying CaM target proteins and CaM sites is an important and ongoing research problem because of the great diversity of conformations it uses in its target interactions. This diversity cannot be captured by a single amino acid sequence motif, but instead CaM-binding sites are commonly divided into four or more motif classes with different sequence characteristics. Historically, CaM-binding sites have been categorized into motifs based on biochemical criteria. Motifs can be either calcium-dependent or calcium-independent based on whether they interact with CaM at basal cellular calcium concentrations (independent) or require elevated calcium (dependent). The 1-10, 1-14 and 1-16 motifs are examples of calcium-dependent motifs and are named to indicate the positions of key hydrophobic residues involved in CaM interaction. Binding sites with the IQ motif are calcium-independent. The figure shows a typical calcium-dependent interaction where the two halves of CaM bind to opposite sides of the target peptide (the four calcium molecules are green spheres).
Calmodulin (CaM) is a calcium-binding protein that is a major transducer of calcium signaling and is a key signaling molecule for multicellular organisms. It has no enzymatic activity of its own but rather acts by binding to and altering the activity on a panel of cellular protein targets at a variety of motifs through binding mechanisms. Its targets are structurally and functionally diverse and participate in a wide range of physiological functions including immune response, muscle contraction and memory formation. Identifying CaM target proteins and CaM sites is an important and ongoing research problem because of the great diversity of conformations it uses in its target interactions. This diversity cannot be captured by a single amino acid sequence motif, but instead CaM-binding sites are commonly divided into four or more motif classes with different sequence characteristics. Historically, CaM-binding sites have been categorized into motifs based on biochemical criteria. Motifs can be either calcium-dependent or calcium-independent based on whether they interact with CaM at basal cellular calcium concentrations (independent) or require elevated calcium (dependent). The 1-10, 1-14 and 1-16 motifs are examples of calcium-dependent motifs and are named to indicate the positions of key hydrophobic residues involved in CaM interaction. Binding sites with the IQ motif are calcium-independent. The figure shows a typical calcium-dependent interaction where the two halves of CaM bind to opposite sides of the target peptide (the four calcium molecules are green spheres).
Motifs are patterns widespread over a group of proteins that are likely to be related by function or may have other biological features in common. Usually, each motif contains a sequence pattern of 3-20 amino acids. Motifs of 3-10 amino acids are considered as short, linear motifs (SLiMs) or minimotifs [10]. SLiMs can have the capacity of encoding a functional interaction in a short sequence, and enrichment in intrinsically disordered regions of proteins. SLiMs should also be able to function independently of their tertiary structure context and their tendency to evolve convergently.
We have studied the role of SLiMs in prediction of CaM-binding proteins and their interactions with other proteins. We have proposed a new method for the prediction of CaM-binding proteins based on both the total and average scores of known and new SLiMs in protein sequences using a new scoring method called sliding window scoring (SWS). Our manually curated dataset contains 194 human CaM-binding proteins and 193 mitochondrial proteins have been obtained and used for testing the proposed model. The motif generation tool. Multiple EM for Motif Elucidation (MEME) has been then used to extract new motifs which combined with support vector machine and random forest classifiers have yielded excellent results. In addition, we have been able to extract new CaM-binding motifs that are currently being studied. The manually curated dataset is available here.
Relevant publications:
- Y. Li, M. Maleki, N. Carruthers, P. Stemmer, A. Ngom, L. Rueda. "The Predictive performance of Short-Linear Motif Features in the Prediction of Calmodulin-Binding Proteins". BMC Bioinformatics. Dataset available here.
- Y. Li, M. Maleki, N.J. Carruthers, L. Rueda, P.M. Stemmer, A. Ngom. "Prediction of Calmodulin Binding Proteins Using Short Linear Motifs". The 5th International Work-Conference on Bioinformatics and Biomedical Engineering (IWBBIO 2017), Granada, Spain, 2017, pp. 107-117.
- Y. Li, L. Rueda, A. Ngom, "Prediction of High-throughput Protein-protein Interactions Using Short Linear Motifs", The GLBIO/CCBC Great Lakes Bioinformatics and the Canadian Computational Biology Conference, 2016, Toronto, Canada. Poster presentation.
The Role of SLiMs in Prediction of PPI
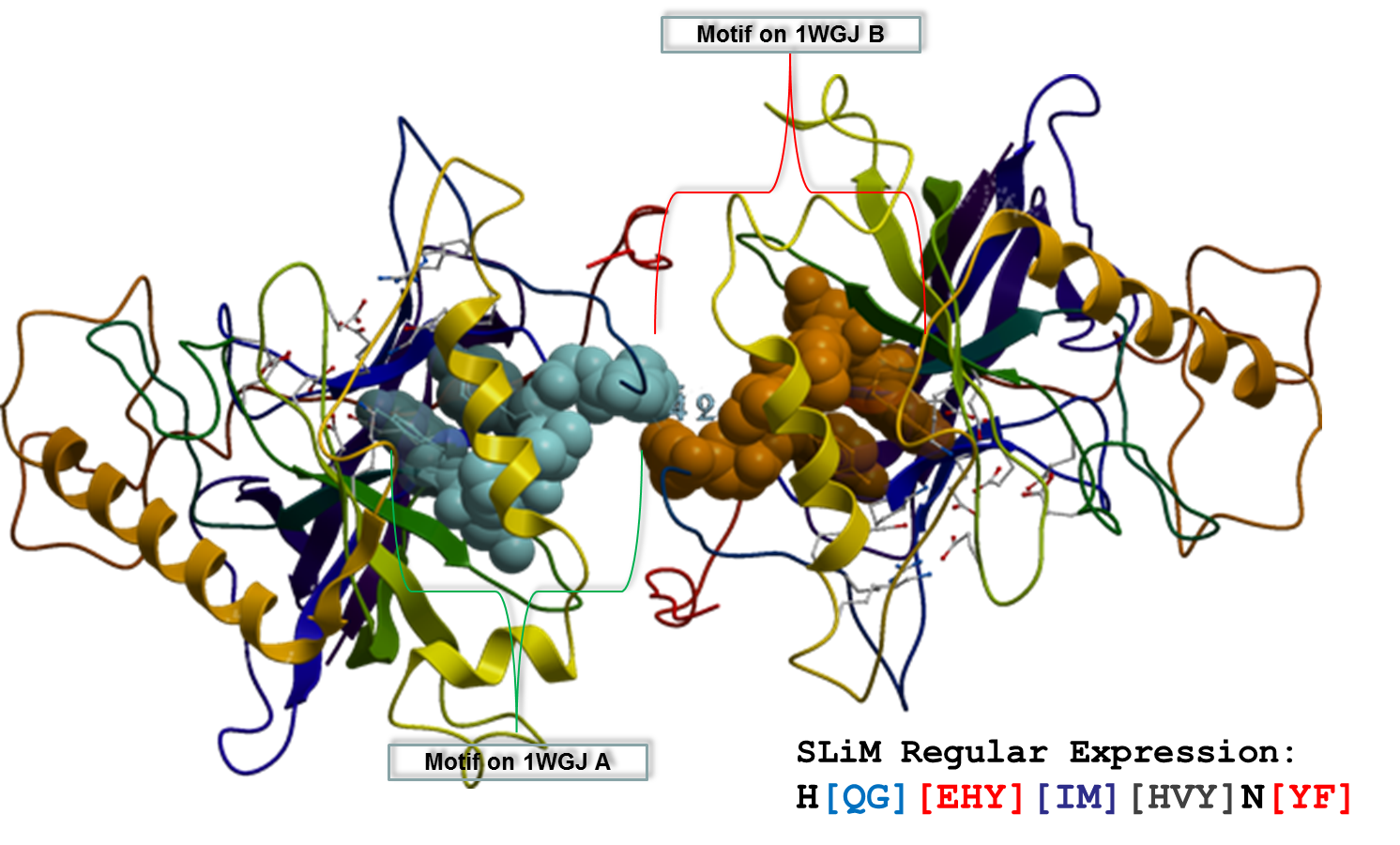 Motifs are patterns widespread over a group of proteins that are likely to be related by function or may have other biological features in common. Usually, each motif contains a sequence pattern of 3-20 amino acids. Motifs of 3-10 amino acids are considered as short, linear motifs (SLiMs) or minimotifs [10]. SLiMs can have the capacity of encoding a functional interaction in a short sequence, and enrichment in intrinsically disordered regions of proteins. SLiMs should also be able to function independently of their tertiary structure context and their tendency to evolve convergently.
Motifs are patterns widespread over a group of proteins that are likely to be related by function or may have other biological features in common. Usually, each motif contains a sequence pattern of 3-20 amino acids. Motifs of 3-10 amino acids are considered as short, linear motifs (SLiMs) or minimotifs [10]. SLiMs can have the capacity of encoding a functional interaction in a short sequence, and enrichment in intrinsically disordered regions of proteins. SLiMs should also be able to function independently of their tertiary structure context and their tendency to evolve convergently.
We have studied the role of SLiMs in protein interactions, especially those contained in the interface of protein complexes (see example in the figure). We have proposed a model that uses SLiMs as properties to predict obligate and non-obligate protein interaction types. Using various classifiers such as k-NN, LDR and SVM on two well-known datasets, we have achieved very high prediction performance, with an increase of at least 7% in accuracy from previous approaches, even better than the structure-based methods, while using only sequence information.
Relevant publications:
- M. Pandit, L. Rueda (2013). Prediction of Biological Protein-protein Interaction Types Using Short, Linear Motifs. ACM Conference on Bioinformatics, Computational Biology and Biomedicine (ACMBCB 2013), Washington, DC, USA, pp. 699-700.
- M. Pandit, L. Rueda (2013). Prediction of Obligate Protein-protein Interactions Using Short, Linear Motifs. 17th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2013), Beijing, China. Poster presentation.
Electrostatic and Desolvation Energies in Prediction of PPI
 Different approaches for prediction of PPI have used a wide variety of physicochemical properties. Of these, we have found that binding free energies are relevant for prediction of obligate complex types. Knowledge-based contact potential that accounts for hydrophobic interactions, self-energy change upon desolvation of charged and polar atom groups, and side-chain entropy loss is defined as desolvation energy. In our earlier work, we have used desolvation energies to predict types of complexes, achieving very good prediction accuracy. We have also used desolvation energies to predict crystal packing and biological complexes.
Different approaches for prediction of PPI have used a wide variety of physicochemical properties. Of these, we have found that binding free energies are relevant for prediction of obligate complex types. Knowledge-based contact potential that accounts for hydrophobic interactions, self-energy change upon desolvation of charged and polar atom groups, and side-chain entropy loss is defined as desolvation energy. In our earlier work, we have used desolvation energies to predict types of complexes, achieving very good prediction accuracy. We have also used desolvation energies to predict crystal packing and biological complexes.
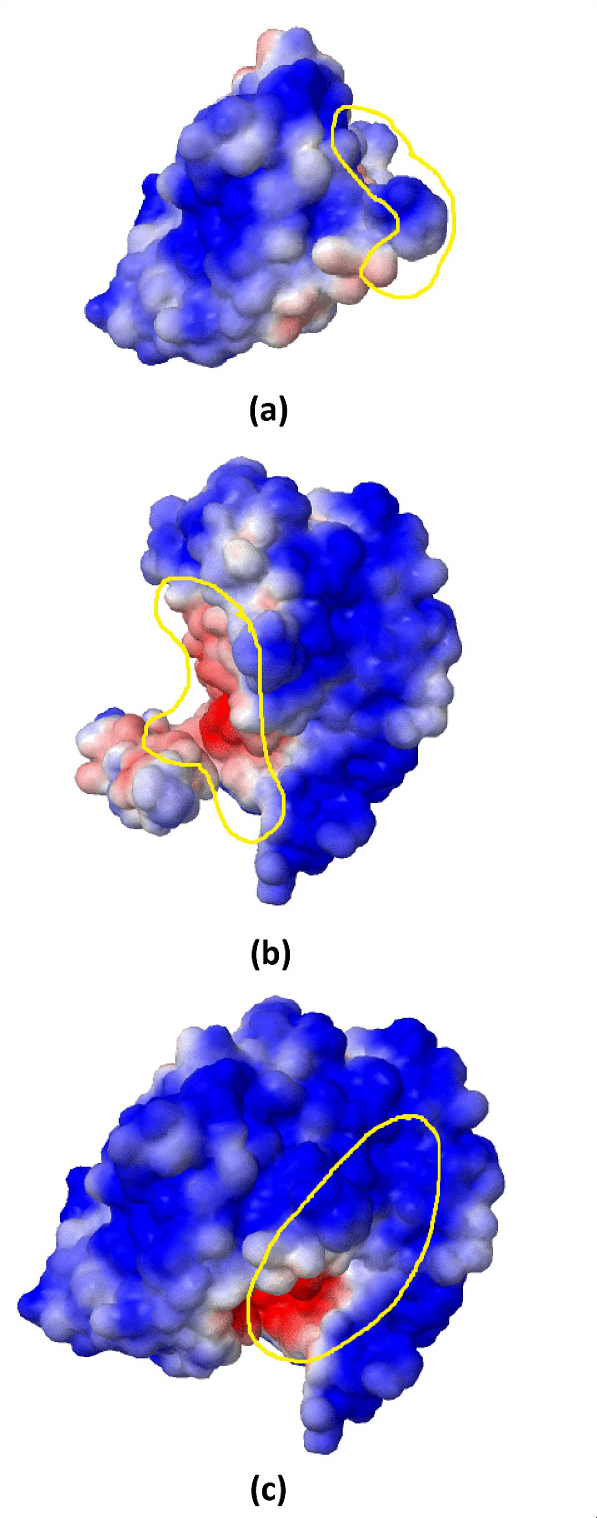
Electrostatic interactions are one of three types of non-covalent interactions, which occur between electrically charged atoms having both positive and negative interactions. Non-covalent interactions are very common between macromolecules such as proteins. Electrostatic energy involves a long-range interaction and can occur between charged atoms of two interacting proteins or two different molecules. Moreover, these interactions can occur between charged atoms on the protein surface and charges in the environment. For example, in an obligate protein complex (see figure) positively charged atoms (red highlighted area in (b)) and negatively charged atoms (blue highlighted area in (a)) show high affinity and hence a stronger interaction. We have proposed a model that uses electrostatic energies of pairs of atom types and amino acids to predict protein complex types. We have computed electrostatic energy values via PDB2PQR and APBS, and used them to predict obligate and non-obligate protein complexes.
Our results on two well-known datasets of obligate and non-obligate complexes confirm that electrostatic energy is an important property for prediction on the basis of all the experimental results, achieving accuracies of over 95%. Furthermore, a comparison performed by changing the distance cutoff demonstrates that the best values for prediction of PPI types using electrostatic energy range from 9Å to 12Å, which show that electrostatic interactions are long-range and cover a broader area in the interface. We have also applied feature selection mechanisms to show that (a) a few pairs of atoms and amino acids are appropriate for prediction, and (b) prediction performance can be improved by eliminating irrelevant and noisy features and selecting the most discriminative ones.
Relevant publications:
- M. Maleki, G. Vasudev, L. Rueda (2013). The role of electrostatic energies in prediction of obligate protein-protein interactions. BMC Proteome Science, 2013, 11(Suppl 1):S11. Work presented at BIBM 2012 (invited talk).
- Md. Aziz, M. Maleki, L. Rueda, M. Raza, S. Banerjee (2011). Prediction of Biological Protein-protein Interactions using Atom-type and Amino Acid Properties. Proteomics, 2011, 11(19):3802–3810. Work presented at CIBCB 2012, BIBM 2011, BIOKDD 2011 and BIBM 2010.
Sequence and Structural Domains in PPI
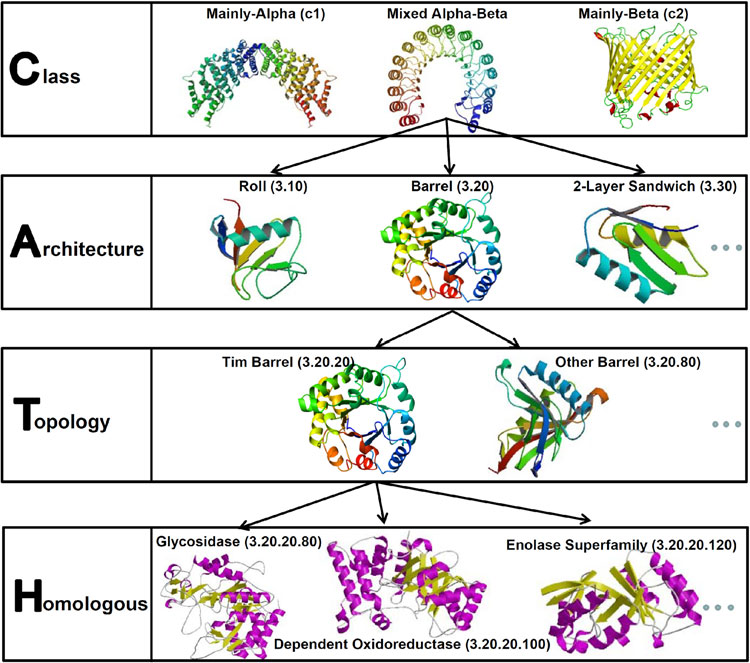 Domains can be considered to be the minimal and fundamental units of proteins. Whether domains are sequence or structural, in most of the cases, they are associated with a specific biological role and act as basic functional units within cells. Previous studies have focused on employing domain knowledge to predict PPI. In our earlier work we proposed a prediction model that uses Pfam domains to predict obligate and non-obligate PPIs. The results demonstrated that desolvation energies are more efficient and powerful than interface area and composition properties for prediction and that homo domain interactions are associated with obligate complexes.
Domains can be considered to be the minimal and fundamental units of proteins. Whether domains are sequence or structural, in most of the cases, they are associated with a specific biological role and act as basic functional units within cells. Previous studies have focused on employing domain knowledge to predict PPI. In our earlier work we proposed a prediction model that uses Pfam domains to predict obligate and non-obligate PPIs. The results demonstrated that desolvation energies are more efficient and powerful than interface area and composition properties for prediction and that homo domain interactions are associated with obligate complexes.
In a recent work we have proposed a model to predict obligate and non-obligate protein interaction types using desolvation energies of structural domains that are present in the interfaces of protein complexes, which are extracted from the CATH database. The prediction is performed using several classifiers on two well-known datasets, which demonstrate that domain-based features of higher levels of CATH, especially level 2, are more powerful and discriminative than features of other levels. The study also concluded that properties taken from different levels of the CATH hierarchy yield higher accuracies than properties taken from each level of the hierarchy separately. Furthermore, analysis of structural properties suggests that domain–domain interactions that have at least a mainly-beta secondary structure in one sub-unit are more informative for predicting obligate and non-obligate PPIs.
Relevant publications:
- M. Maleki, M. Hall, L. Rueda (2013). The Role of Structural Domains in Prediction of Protein-protein Interaction Types. Network Modeling Analysis in Health Informatics and Bioinformatics. DOI 10.1007/s13721-013-0043-9. Work presented at ACM-BCB 2012, CIBCB 2012, BIBM 2011 and BIOKDD 2011.
Dynamics and Proteotranscriptomics
We are currently investigating the following emerging topics associated with the dynamics in the interactome and its relationships with transcriptomics.
- Dynamics of protein interactions by means of studying temporal associations of proteins into transient/permanent or obligate/non-obligate complexes.
- Data integration of interactomics with transcriptomics by finding relationships between gene expression and alternative splicing and protein-protein interactions. Applications to breast and prostate cancer are being pursued. In one of the projects (see also Transcriptomics) we are currently investigating the protein variants or isoforms found in RNA-seq data and which are associated with alternative splicing in prostate cancer.
- Interactomics in oral fluids, in particular, the main aspects of salivary protein-protein interactions.